|
|
|
|
|
|
Katten aids
HCM:
Hypertrophic Cardiomyopathy in Cats,
Report on Hypertrophic Cardiomyopathy from the First International Feline Genetic Disease Conference held June 25-28, 1998 at the University of Pennsylvania, Philadelphia, PA (Updated with Grant Progress Report)
by Susan Little, DVM, Diplomate ABVP (Feline)
The two primary researchers into heritable hypertrophic cardiomyopathy (HCM) in cats are Dr. Mark Kittleson of the University of California at Davis and Dr. Kathryn Meurs of The Ohio State University.
Both researchers were present at the Feline Genetic Disease Conference for presentations on HCM and question and answer sessions with both veterinarians and breeders.
Dr. Kittleson's presentation began with an overview of HCM in humans, where it is known to be inherited in an autosomal dominant fashion and has an adult onset.
While there are currently over 100 mutations identified in seven different genes that can be responsible for HCM in people, the clinical signs and progress of the disease are often the same. The most common genetic defect involves a mutation in a gene that codes for the structure of an integral muscle protein (the beta-myosin heavy chain) in the individual heart muscle cell. Myosin is a protein that makes up about 65% of all the protein in a muscle cell. It consists of long chains of polypeptide components that are joined to each other by side chains. Myosin is one of the proteins responsible for contraction of heart muscle.
In cats, HCM is the most common cardiac disease. Most cats are middle-aged at the onset of their clinical symptoms, but patients as young as one year old and as old as 13 years have been identified. The average cat with severe HCM and heart failure lives for only several months despite therapy.
The first investigations into HCM Cats started when Marcia Munro, a private cat owner from Connecticut, contacted Dr. Kittleson about her affected Maine Coon cat and the related cats she had identified with HCM. All the cats from the cattery of origin were ultrasounded, and one unaffected male was bred to three affected females to start a research colony.
In the research colony, ultrasounds were done starting at three to six months of age and were done every four to eight months afterward. Breeding records showed that HCM was inherited as an autosomal dominant trait in this colony. Breeding an affected cat to any other cat always produced at least one affected kitten in the litter. Equal numbers of males and females are affected. In Dr. Kittleson's colony, no cats with HCM were identified before one year of age when test breedings were done between affected cats and non-affected cats. The disease was often evident by two years of age and could be very severe by two to four years of age. Males seemed to have more severe disease and were affected earlier. When affected cats were bred to other affected cats, however, HCM appeared as early as three months of age; severe disease could occur by six to 18 months of age and males and females were affected in the same way. In the general cat population, cats with severe HCM will usually die of their disease. More males die of HCM and there may be no symptoms until the sudden death of the cat.
Dr. Kittleson identified certain characteristics of the HCM in cats that can be seen on ultrasound. Two changes in particular, called systolic interior motion of the mitral valve and papillary muscle hypertrophy, are commonly seen in Cats with HCM. Dr. Meurs investigated the possibility that the genetic defect in HCM in cats is similar to that in people (i.e., it is in the beta-myosin heavy chain).
Her studies found, however, that while cats often had genetic differences in the beta-myosin heavy chain, they were not differences that affected the production of the protein. Attention has now turned to other genes that make proteins that are part of the contractile element (the assembled proteins responsible for contraction) in heart muscle to determine if one of them is responsible for familial feline HCM. These include the myosin light chains and myosin binding protein C genes. In humans there is a rare form of HCM that involves a defect in the myosin light chains that produces primarily papillary muscle hypertrophy.
Dr. Kittleson has also looked at HCM in other breeds, including the American Shorthair. The disease in this breed is also inherited as an autosomal dominant trait; however, the disease commonly is not as severe.
Dr. Kittleson's recommendations for screening cats for breeding depend on the sex of the cat. He recommends that male cats be screened at two years of age or older (because they often have earlier and more severe disease) and female cats be screened later, perhaps at three to four years of age. He stressed that it is very important to have an ultrasonographer who is familiar with the particular components of HCM in s as the early changes (such as left papillary muscle hypertrophy) might be missed by the uninformed person.
Resources
Dr. Kittleson's work has been presented at recent veterinary conferences in addition to the Feline Genetic Disease Conference:
Meurs K, Kittleson MD, Towbin J, Ware W. Familial systolic anterior motion of the mitral valve and/or hypertrophic cardiomyopathy is apparently inherited as an autosomal dominant in a family of American Shorthair cats.
Proceedings of the 15th American College of Veterinary Internal Medicine Forum, Lake Buena Vista, FL, 1997.
Kittleson MD, Meurs KM, Kittleson J, Munro M, Liu S, Towbin JA. Heritable characteristics, phenotypic expression, and natural history of hypertrophic cardiomyopathy in cats. Proceedings of the 16th American College of Veterinary Internal Medicine Forum, San Diego, CA, 1998.
The research has also been published in the following journals/books:
Kittleson MD. Development and progression of inherited hypertrophic cardiomyopathy in cats (abstract).
J Vet Internal Med, Vol 10, No 3, p 165, 1996.
Meurs K, Kittleson MD, Towbin J, Ware W. Familial systolic anterior motion of the mitral valve and/or hypertrophic cardiomyopathy is apparently inherited as an autosomal dominant in a family of American Shorthair cats.
J Vet Internal Med, Vol 11, No 2, p 138, 1997.
Kittleson MD, Kienle RD. Small Animal Cardiovascular Medicine, Mosby, St. Louis, MO, 1998.
AUTOSOMAL DOMINANT GENE
All chromosomes are present in pairs, except for the sex chromosome (X and Y). The chromosomes other than X and Y are called autosomes. A genetic trait that is inherited on the non-sex chromosomes is called autosomal. A genetic trait that is inherited on the sex chromosomes is termed "sex-linked." Most traits are autosomal, including HCM in cats.
The chromosomes contain thousands of genes. Since each autosome is paired, genes on those chromosomes are present in pairs. Each time a body cell divides, the genes multiply with a process of self-copying which is highly accurate. Occasionally, an inexact copy of a gene is made and this is termed a "mutant." Variations from normal types or conditions usually occur as the result of mutations, and this is the case for HCM.
When a disease is caused by a mutation in a gene that codes for a structural or functional protein, a mutation in only one copy of the gene ("allele") can produce disease since a full complement of the protein is needed for the affected organ to function normally. When only one allele (either from the mother or father) needs to be affected to cause disease, it is an autosomal dominant trait. Mutations in genes that code for enzymes usually need both alleles (one each from the mother and father) affected since enzyme systems generally have to be suppressed to less than 10% of their normal level to produce an effect (in this case a disease). This is called an autosomal recessive disease. Only one copy of the mutant gene (one allele) is needed to produce HCM in a cat.
Basic Science Reports
Background—A naturally occurring animal model of familial hypertrophic cardiomyopathy (FHCM) is lacking. We identified a family of cats with HCM and developed a colony to determine mode of inheritance, phenotypic expression, and natural history of the disease.
Methods and Results—A proband was identified, and related cats were bred to produce a colony. Affected and unaffected cats were bred to determine the mode of inheritance. Echocardiography was used to identify affected offspring and determine phenotypic expression. Echocardiograms were repeated serially to determine the natural history of the disease.
Of 22 offspring from breeding affected to unaffected cats, 12 (55%) were affected. When affected cats were bred to affected cats, 4 (45%) of the 9 were affected, 2 (22%) unaffected, and 3 (33%) stillborn.
Findings were consistent with an autosomal dominant mode of inheritance with 100% penetrance, with the stillborns representing lethal homozygotes that died in utero. Affected cats usually did not have phenotypic evidence of HCM before 6 months of age, developed HCM during adolescence, and developed severe HCM during young adulthood. Papillary muscle hypertrophy that produced midcavitary obstruction and systolic anterior motion of the mitral valve was the most consistent manifestation of HCM.
Cats died suddenly (n=5) or of heart failure (n=3). Histopathology of the myocardium revealed myocardial fiber disarray, intramural coronary arteriosclerosis, and interstitial fibrosis.
Conclusions—HCM in this family of cats closely resembles the human form of FHCM and should prove a valuable tool for studying the gross, cellular, and molecular pathophysiology of the disease.
Key Words:
• cardiomyopathy
• hypertrophy
• heart diseases
• genetics
Familial hypertrophic cardiomyopathy (FHCM) is a common hereditary human disease caused by mutations in 7 genes that encode for sarcomeric proteins.1 HCM has been recognized as a common cause of heart failure, sudden death, and systemic thromboembolism in domestic cats since the 1970s.
2 3 4 Feline HCM has previously been touted as a model of the human disease.
2 4 However, these cats are client-owned and generally unavailable for detailed study, only 40% have histological evidence of myocardial fiber disarray, and no heritable basis for the disease has been described.4 5
This report describes a colony of cats with a heritable form of HCM that mimics human FHCM. It is characterized by moderate to severe papillary muscle and left ventricular (LV) concentric hypertrophy, systolic anterior motion (SAM) of the mitral valve, and the typical histopathological features seen in human FHCM. The natural history of the disease mimics that seen in humans, and it is inherited as an autosomal dominant trait.
This study was conducted in accordance with the "Position of the American Heart Association on Research Animal Use" and under the guidelines of the Animal Care and Use Committee of the University of California at Davis.
Animal Procurement
Initially, a proband and several related cats with HCM were identified. Echocardiograms were performed on 35 cats from the cattery of origin. Seven were diagnosed with HCM (affected cats). One unaffected male (the father of the proband) was bred to 3 affected females (the proband's mother, the mother's sister, and a sister of the proband). The breeding between this male and 2 of the affected females had produced affected kittens previously. Eight kittens from these breedings and 1 affected female were obtained to start a breeding colony.
Determination of Inheritance Pattern
Heritable traits were determined through planned breedings between affected and unaffected colony cats (group 1), affected and affected cats (group 2), and unaffected and unaffected cats (group 3). Affected cats were identified through careful and repeated echocardiographic examinations, as described below. Relationship data were collated and analyzed to determine the mode of inheritance.
Determination of Phenotypic Expression
Standard feline echocardiographic studies were performed with an Acuson 128XP/10 ultrasound machine and a 7-MHz transducer. The cats were unsedated and restrained in right and left lateral recumbency on a Plexiglas table. The transducer was introduced from below through a medium-sized rectangular hole in the table to maximize the echocardiographic window. Standard right parasternal long-axis and short-axis views plus left apical and left cranial views were examined.6 Papillary muscle and left atrial sizes were judged subjectively by 1 investigator (M.D.K.) as normal or mildly, moderately, or severely enlarged. Measurements of diastolic LV wall thickness were made from the 2-dimensional image.
Cats were suspected of having HCM when moderate papillary muscle hypertrophy or SAM was present without LV wall hypertrophy.
Cats were definitively diagnosed with HCM when severe papillary muscle hypertrophy was present or a region of the LV wall or the entire wall of the LV was 6 mm thick.4 The disease was considered severe when papillary muscle hypertrophy was severe or at least 1 LV wall was 7 mm thick.
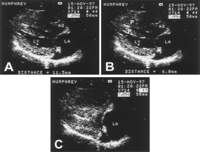
A, Short-axis right parasternal echocardiogram from a normal cat. Papillary muscles (arrowheads) are normal. B, Echocardiogram from a group 1 affected cat at 4.5 months of age. LV and papillary muscles are normal. C, At 11 months, mild to moderate papillary muscle enlargement is present. D, At 17 months, papillary muscles are severely enlarged and LV free wall is severely thickened (9.5 mm).
The presence or absence of SAM was recorded. Cats with SAM had a grade II to IV/VI left apical systolic heart murmur that was dynamic (loud when the cat was excited, softer when relaxed). SAM was most reliably identified from a right parasternal long-axis view of the LV, LV outflow tract (LVOT), and aorta; a cine loop; and the motion of the anterior mitral valve leaflet with respect to the LVOT and interventricular septum from the early part of systole through end systole (Figure 3A and 3B). On color flow Doppler echocardiography, 2 turbulent jets, 1 of dynamic subaortic stenosis (SAS) and the other of mitral regurgitation, were seen originating from the region where the anterior mitral valve leaflet encroached on the LVOT (Figure 3C). The peak velocity of the dynamic SAS jet was obtained with continuous-wave Doppler echocardiography and converted to a pressure gradient by use of the modified Bernoulli equation.
Natural History
Echocardiograms were first performed at 3 to 6 months of age. The cats underwent ultrasonography every 4 to 8 months to identify cats that developed HCM and to document the progression of the disease. The ages at which HCM was diagnosed and at which moderate to severe HCM was definitively diagnosed were recorded. Congestive heart failure was diagnosed when radiographic evidence of pulmonary edema or pleural effusion was present. Congestive heart failure was treated until the cat became refractory to therapy. Sudden death was defined as a witnessed event of sudden death in a cat or as finding a cat with HCM dead but without pathological evidence of severe heart failure or systemic disease. The age and mode of death were recorded.
Pathological Examination
Hearts from cats that died were trimmed, weighed, and placed in formalin. Maximal septal and LV free wall measurements were obtained. Sections of the LV free wall and septum were cut in a plane perpendicular to the long axis of the LV, and the microscopic abnormalities were categorized and quantified as reported previously.5 7
Statistical Analysis
The differences in occurrence and progression of disease and in sex between groups 1 and 2 were analyzed by Student's unpaired t tests. The null hypothesis was discarded at P.
Inheritance Pattern and Natural History
Forty kittens in 10 litters were produced, of which 33 were included in the pedigree. All breedings produced live kittens. Stillborn kittens were present in some litters and were included in the pedigree. Stillborn kittens were formed but not totally developed, were hairless, had undergone autolysis, and had obviously died in utero. Kittens with omphaloceles (n=5) or pectus excavatum (n=2) were euthanized or died soon after birth and were not included in the pedigree. One cat with a surgically corrected portosystemic shunt and a cat with genu recurvatum were maintained and included in the pedigree.
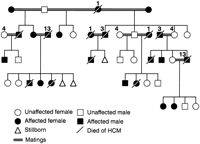
Unaffected x Affected Breedings (Group 1)
Seven litters and 28 kittens were the result of mating an affected cat with an unaffected cat (Figure 1). Twenty-two kittens survived. Five kittens had omphaloceles and 1 had pectus excavatum. No stillborn fetuses were present.
Twelve of the 22 surviving kittens developed HCM (7 males and 5 females). No evidence of HCM was present in any group 1 cat before 6 months of age. Papillary muscle enlargement, usually along with SAM of the mitral valve, developed first, most commonly between 9 and 21 months of age (16±5 months; Figure 2). One female did not have any change until 38 months of age. The age at which LV wall thickening or papillary muscle hypertrophy became moderate to severe was between 13 and 48 months (24±13 months). The wall thickening was often localized primarily to the papillary muscles and the posterolateral free wall, between the papillary muscles (Figures 2D, 4, and 5). The disease became severe earlier in males (n=5; 19±6 months) than in females (n=5; 29±13 months).
Five group 1 cats died of HCM: 2 of heart failure and 3 suddenly. Four of the 5 were males. The age at which heart failure was first noted was 33±10 months. The age of death (sudden or heart failure) was 32±12 months.
Affected x Affected Matings (Group 2)
Two litters containing 10 kittens or stillborn fetuses resulted from breeding 2 affected cats. One litter had 3 kittens: 1 with pectus excavatum, 1 stillborn fetus, and 1 male that developed HCM. The other litter had 5 kittens that survived and 2 stillborn fetuses. One male and 2 females developed HCM, and 2 females did not develop HCM (Figure 1). Both males and 2 of the 4 females developed severe HCM in group 2. Disease in this group appeared significantly earlier and progressed more rapidly than in group 1. The echocardiographic findings were suggestive of HCM when cats were 4 to 6 months of age (5±1 months). The disease was severe by 7 to 12 months of age (9±2 months). All 4 affected cats developed heart failure and were started on heart failure therapy between 18 and 24 months of age (20±3 months). The 2 males died suddenly at 18 and 25 months of age (Figure 6). One female died of heart failure at 25 months of age. The other female is alive (31 months old). Age of death was 23±4 months. The ages at which HCM was diagnosed, at which HCM was severe, and at which heart failure was noted were significantly less for group 2 than for group 1.
Unaffected x Unaffected Matings (Group 3)
Breeding one unaffected to another unaffected cat produced 2 unaffected cats. Both survived and were 3 years old at the time the study terminated.
Assessment of Inheritance Pattern
The following findings are compatible with an autosomal dominant mode of inheritance with 100% penetrance. Whenever an affected cat was bred to another cat, at least 1 affected cat was produced and successive generations were affected (Figure 1). Nine affected cats were male and 7 female (no sex predilection). Three females and 4 males were the affected parent in group 1 (males and females were equally likely to transmit the disease). Several instances of male-to-male transmission occurred. Of the 22 kittens born from affected (A/a) to unaffected (a/a) matings (group 1), 12 (55%) developed HCM (A/a). Four of the 9 kittens (45%) produced from mating affected (A/a) to affected (A/a) cats (group 2) developed HCM (A/a) and 2 (22%; a/a) did not. Three stillborn fetuses (33%) (assumed to be lethal homozygotes: A/A) were produced from these breedings. Neither group 3 cat developed HCM.
Phenotypic Expression
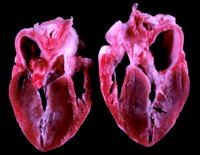
Papillary muscle hypertrophy was the predominant and most consistent abnormality noted. It produced midventricular obstruction in some cats (Figures 5B and 7). Separating the hypertrophied papillary muscles from adjacent hypertrophied myocardium was sometimes difficult, but often a clear separation could be identified (Figure 4). The hypertrophy, however, was often not confined to the papillary muscles. For example, hypertrophy was often noted at the basilar portion of the interventricular septum, a region remote from any papillary muscle influence.
LV wall thickness achieved a maximum and then stabilized in all cats, as it does in humans.8 Left atrial size was normal in cats with mild to moderate hypertrophy and when severe hypertrophy was first identified. Left atrial size then increased in the cats with severe hypertrophy. The exception to this was 1 cat who had 1 massively hypertrophied papillary muscle, no wall hypertrophy, and no left atrial enlargement for 4 years.
SAM was identified in most cats (13 of 16 cats) from the time papillary muscle enlargement was noted. In 2 cats, the SAM disappeared after LV hypertrophy developed. The blood flow velocity across the region of dynamic SAS was highly variable within the population, and within each cat, it was dependent on the level of excitement. An attempt was made to excite each cat to increase flow velocity at the time of examination, but these maneuvers were variably successful. The maximum pressure gradient ranged from 10 to 92 mm Hg (51±26 mm Hg). Cats with moderate to severe disease also had end-systolic cavity obliteration in the region of the LV cavity that contained the papillary muscles (Figure 4C).
Pathological Examination
Of the 8 animals that died, 7 had severe HCM. The heart weight was 29 to 37 g (normal is < 20 g) in these cats.9 10 11 The remaining cat died suddenly before exhibiting echocardiographic evidence of HCM (heart weight, 24 g). The ratio of heart weight to body weight was severely increased in the cats with severe HCM (8.1 to 10.6 g/kg; normal, 3.0 to 5.0 g/kg) and was mildly increased (6.0 g/kg) for the cat that died suddenly without severe HCM. Maximal ventricular free wall thickness was significantly increased to 10.7±1.3 mm (normal, 5.0±0.2 mm) and interventricular septal thickness to 9.6±0.7 mm (normal, 5.0±0.3 mm).11 Because cardiac muscle often undergoes postmortem contracture, the wall thicknesses at postmortem examination were often greater than those measured by echocardiography. Hypertrophy consistently involved the papillary muscles along with substantial portions of the LV walls. Moderate to marked (>5% of the tissue section) myofiber disarray (predominantly type I) of the interventricular septum and LV free wall, moderate to severe intramural coronary arteriosclerosis, and moderate to severe interstitial fibrosis were present in all cats.
We have identified a familial form of HCM in cats that closely mimics FHCM in humans. Although naturally occurring HCM has been described in humans, dogs, cats, and pigs, this is only the second species in which a naturally occurring and heritable form of HCM has been identified.12 13 14 15 However, a heritable basis for HCM in pigs is suspected.
HCM in these cats appears to be inherited as an autosomal dominant trait, as it is in human FCHM, with penetrance that increases to 100% in adulthood. Although penetrance may not be 100% in some human families with FHCM, often it is complete, as in our cats.
HCM in our colony of cats has most of the common morphological characteristics present in human FHCM. It is characterized by gross LV wall thickening, dynamic LVOT obstruction, myocardial fiber disarray, small coronary artery disease, and myocardial fibrosis. The one difference is that the LV wall thickening is more commonly confined to or more severe in the papillary muscles and LV free wall than in the interventricular septum. Asymmetric hypertrophy of the free wall has also been identified in a murine transgenic model of HCM in which the -myosin heavy chain gene (MHCG) contains a mutation commonly identified in humans with HCM.20 Consequently, this finding may be purely a species difference in the expression of the disease. However, a rare form of HCM involving primarily the papillary muscles exists in humans and is caused by mutations in either the essential or regulatory myosin light chains.21 It is characterized by midventricular obstruction due to the papillary muscle hypertrophy.
The natural history of FHCM in cats is similar to human FHCM, in which the disease commonly becomes evident during childhood or adolescence and progressively worsens during young adulthood.22 In our colony, most cats first had definitive evidence of HCM between 8 and 24 months of age. The disease usually reached its nadir between 1.5 and 3 years of age. Cats generally become mature adults between 12 and 18 months of age, so these periods correspond to adolescence and young adulthood. In a few female cats, however, the disease was not evident until 3 years of age. The progressive nature of the disease in cats is also similar to that seen in transgenic mice with a -MHCG mutation in which HCM is not demonstrable at 5 weeks old but is by 15 weeks.20
Sudden cardiac death is another feature common to both humans and cats with HCM.23 Five of our 16 affected cats died suddenly. One 16-month-old cat without severe hypertrophy but with myocardial fiber disarray died suddenly, as has been described in humans.
FHCM is caused by sarcomeric protein gene mutations in humans.1 Because the clinical, pathological, and heritable characteristics of our feline model are similar to those of human FHCM, it is possible that a sarcomeric gene mutation causes FHCM in this model. It is this targeted group of genes that we are currently screening for mutations. If a mutation could be identified, the model could then be used to test diagnostic methods and therapeutic strategies, including means to prevent the progression and complications of myocardial hypertrophy and to examine gene therapy.
As in transgenic mice, HCM in our cats progressed to severe HCM and death more commonly in males than in females.20 In 1 study, male transgenic mice were affected at an earlier age than females. In addition, left atrial enlargement was present in 7 of 11 mutant males but only 2 of 12 female 15-week-old mice and in all 7 males but only 6 of 18 female 30-week-old mice. More male mice also had myocardial fiber disarray. It appears that male influences in some way modify the phenotypic expression of HCM. This could occur through modulation at the gene level or could be due to known sex differences in myocardial performance.
In the pedigree analysis, the pattern of inheritance was consistent with an autosomal dominant trait when the stillborn fetuses were considered lethal homozygotes. Only a few examples of human homozygous individuals who have lived exist in the literature.26 27 In contrast, mice that are homozygous for an -MHCG mutation consistently die before 7 days old.25 It is well recognized that mutations on the same gene, such as the ß-MHCG, alter the severity and age of onset of the disease and result in different prognoses.28 Because the type of mutation in a heterozygote determines longevity, it is reasonable to assume that the type of mutation in a homozygote could also influence survivability. If a mutation of a sarcomeric protein is involved in cats with HCM, it clearly results in malignant disease. Consequently, one would expect to see markedly reduced survival in cats homozygous for the mutation.
The expressivity of the disease in our colony was influenced by the type of breeding. When affected cats were bred to affected cats, their offspring developed HCM at an earlier age, always developed severe disease, and developed severe disease at an earlier age. One explanation is that these individuals were the homozygotes instead of the stillborn fetuses. If this were true, all of the affected cats in group 2 were homozygotes. This is statistically improbable. Another possible explanation is that modifying factors were altered by breeding affected to affected cats. For example, the extent of hypertrophy in humans with HCM is influenced by the angiotensin I converting enzyme insertion/deletion (I/D) polymorphism.29 Individuals with the DD genotype have severe hypertrophy, whereas individuals with the ID or II genotype have mild or no hypertrophy. It is possible that in our cats, this or another polymorphism was encountered in these 2 breedings by chance or that a polymorphism or other modifying factors are coinherited with the mutation responsible for HCM in these cats.
Although the papillary muscles were most consistently and severely hypertrophied, myocardial fiber disarray was often also found in the LV free wall and interventricular septum. A previous study in humans with HCM has shown that myocardial fiber disarray is not confined to the severely thickened regions of the LV.
We conclude that HCM in cats may be an appropriate model of human disease. Access to this model could benefit investigators interested in examining the gross, cellular, and molecular pathophysiology of HCM. The ability to selectively breed affected to unaffected individuals could provide clues regarding the natural history of the disease in humans, especially during the emerging phase of the disease in adolescence, and might provide clues about why the disease is not expressed at birth. It could also allow investigators to study the offspring of matings of affected to affected individuals, something that occurs only rarely in humans. The model might also be useful to determine why the hypertrophy in HCM is so often asymmetrical instead of symmetrical, as it is in a pressure overload.
In conclusion, HCM in cats mimics the hereditary aspects, phenotypic expression, natural history, and pathological characteristics of the human familial form of the disease. Consequently, it is likely that FHCM in cats is caused by a mutation identified in humans with HCM. This animal model of FHCM may be a valuable tool for studying the gross, cellular, and molecular pathophysiology of the disease.
Acknowledgments
This study was supported by grants from the Winn Feline Foundation, the Center for Companion Animal Health at UC Davis, the NIH, NHLBI (HL-03236-05), and the Texas Children's Hospital Foundation Chair in Pediatric Cardiology Research.

Terug naar medisch
| |